

Ein perfektes Modell, um Myotonien zu erforschen
Seit mehreren Jahren experimentieren Manfred Grabner und Anamika Dayal vom Institut für Pharmakologie der Medizin Uni Innsbruck an einem von ihnen entwickelten Kalziumkanalmodell. Im Rahmen von zwei neuen Studien konnten sie nun den molekularen Mechanismus der Kalzium-Nichtleitfähigkeit im Skelettmuskel dieses Tiermodells identifizieren und schließlich dessen Eignung zur Erforschung seltener Muskelerregungsstörungen endgültig belegen.
Betroffene greifen nach einem Wasserglas, auf halber Höhe zum Mund verkrampfen sich die Fingermuskeln – und genauso plötzlich lösen sie sich wieder. Das Glas fällt aus der Hand. Eine krampfartige Muskelkontraktion und eine unmittelbar darauffolgende, vorübergehende Muskelerschlaffung sind Symptome einer seltenen, erblichen Myotonie, der Myotonia congenita (Becker Krankheit). Bereits 2017 haben Manfred Grabner und Anamika Dayal vom Institut für Pharmakologie der Medizinischen Universität Innsbruck ein Kalziumkanal-Tiermodell vorgestellt und publiziert. Nun ist es dem Forscher-Duo gelungen, sowohl grundlegende biophysikalische Mechanismen ihres Modells zu klären, als auch dessen Überlegenheit gegenüber einem anderen Kalziumkanalmodell zu beweisen. Zwei wissenschaftliche Arbeiten wurden unlängst im renommierten Fachjournal eLife veröffentlicht. „Die Hauptbotschaft ist, dass unser Modell sehr gut geeignet ist, um Krankheiten des Skelettmuskels zu studieren, bei denen Kalzium eine Rolle spielt“, sagt Dayal.
Bereits 2010 hatten die beiden Wissenschafter entdeckt, dass bei Zebrafischen und anderen höheren Knochenfischen der Kalziumstrom durch den Dihydropyridinrezeptor (DHPR) – einer der beiden Kalziumkanäle, die bei der Muskelkontraktion zusammenarbeiten– evolutionär stillgelegt worden war. „Das Kalzium wurde blockiert, um die Muskeln schneller zu machen. Das haben wir herausgearbeitet und die entsprechende zentrale Porenmutation N6117D in das DHPR Gen der Maus transferiert“, so Grabner. Die Innsbrucker Forscher fanden damit eine Möglichkeit, um den Kalziumdurchfluss durch den DHPR komplett zu stoppen, „darüber hinaus entwickelte sich die Maus jedoch völlig gesund mit komplett intakten Muskeln.“ Damit hatten die beiden ein seit über fünfzig Jahren studiertes Rätsel gelöst. „Niemand wusste bis dahin, ob das Kalzium, das über den Dihydropyridinrezeptor einströmte, überhaupt gebraucht wird, oder nicht. Der Kalziumstrom durch den DHPR ist ein evolutionäres Relikt“, schildert Grabner.
Das Vergleichsmodell zur Innsbrucker (non-conducting) ncDHPR Maus, die ebenfalls nicht Kalzium-leitende EIIIK-Maus einer US-Gruppe, zeigte unterdessen Stoffwechselerscheinungen wie u.a. Adipositas und vorzeitige Muskelerschöpfung. „Der DHPR des Vergleichsmodells war allerdings weiterhin durchlässig für andere Ionen, wie Natrium und Kalium, was zu einer Überladung und dadurch zu Problemen führte. Unser System ist überhaupt nicht durchlässig, weder von extra- noch intrazellulärer Seite“, schildert Dayal. Selbst unter experimentellen Bedingungen von denen bekannt ist, dass sie die Kalzium-Leitfähigkeit von Wildtyp-DHPR-Kanälen erhöhen, konnten bei den ncDHPR-Mäusen weder nachweisbare Kalzium- noch Natrium- oder Kalium-Einwärtsströme festgestellt werden, was auf die hohe Ionendichtheit des ncDHPR-Kanals hinweist. Die biophysikalische Erklärung: Die zentrale Porenmutation (N617D) führt in der ncDHPR-Maus eine zweite hochaffine Kalzium- Bindungsstelle in die DHPR Kanalpore ein, die vom kanonischen hochaffinen Kalzium-Selektivitätsfilter weit genug räumlich getrennt ist. Kalzium Ionen die an die beiden Bindungsstellen andocken zeigen dadurch keine signifikante elektrostatische Abstoßung voneinander mehr. „Durch diese elektrostatische Abdichtung des Kanals wird der Eintritt weiterer Ionen verhindert, und der Strom durch den DHPR kommt zum Erliegen“, veranschaulicht Dayal den Mechanismus.
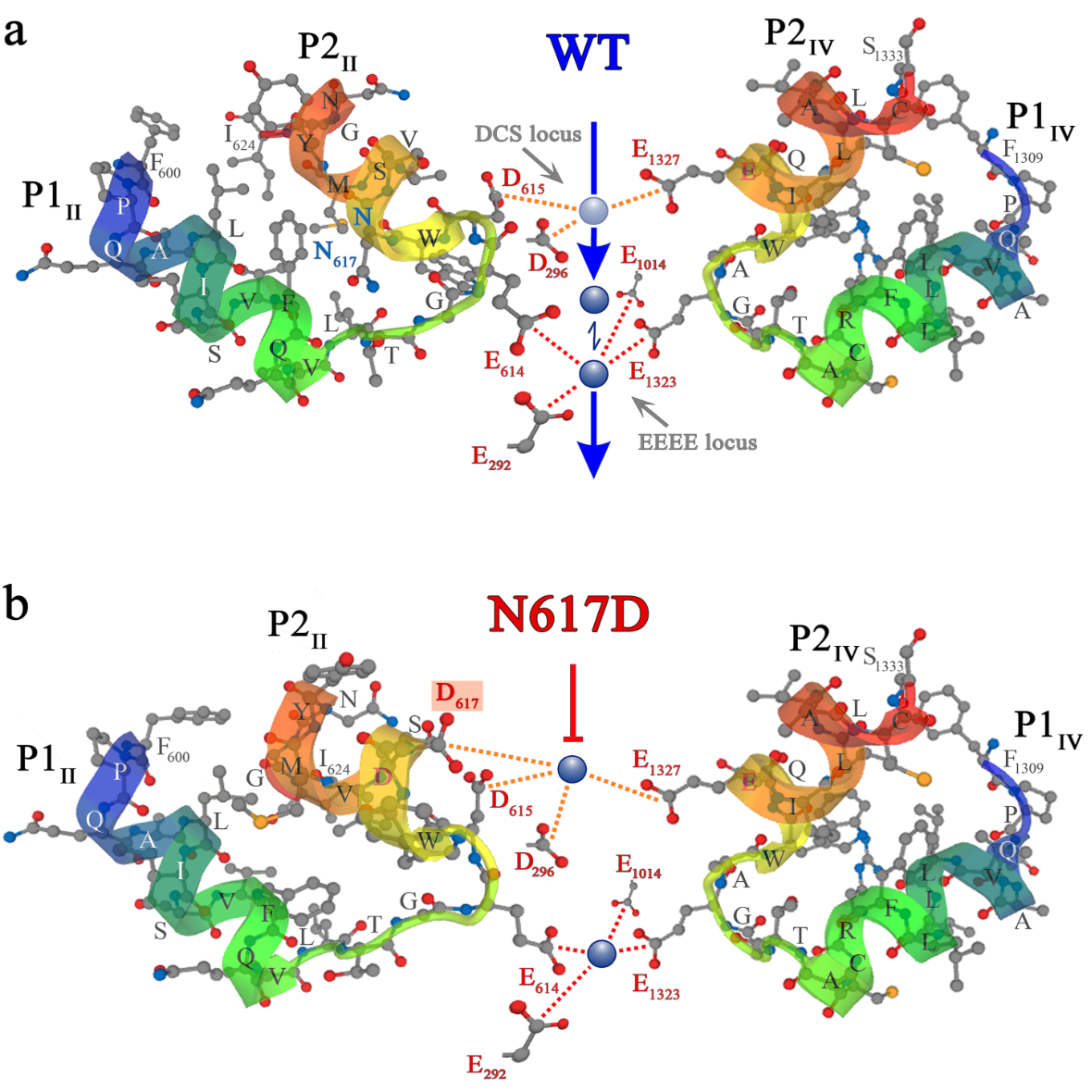 Grafik: Das in eLife veröffentlichte Modell zeigt den Kalziumeinstrom durch den DHPR in der Wildtyp-Maus (o.) sowie den Blockademechanismus im ncDHPR-Mausmodell.
Grafik: Das in eLife veröffentlichte Modell zeigt den Kalziumeinstrom durch den DHPR in der Wildtyp-Maus (o.) sowie den Blockademechanismus im ncDHPR-Mausmodell.
Mit Unterstützung des ncDHPR-Mausmodells konnte nun in der mit dem Labor von Mark M. Rich an der Wright State University in Dayton (US-Staat Ohio) durchgeführten Untersuchung der rezessiven Myotonia congenita der Mechanismus der vorübergehenden Lähmung aufgeklärt werden. So zeigte sich unter anderem, dass die pharmakologisch oder genetisch induzierte Muskelschwäche in der Innsbrucker ncDHPR Maus deutlich verkürzt war – und, dass deshalb „die vorübergehende Lähmung durch Blockade des DHPR Kalziumeintritts frühzeitig terminiert werden kann“, sagt Grabner.
Manfred Grabner und Anamika Dayal sind überzeugt davon, dass ihre Erkenntnisse gleichermaßen für Muskelphysiologen und für Mediziner, die sich mit Myotonia congenita und anderen Muskelerregungsstörungen beschäftigen, interessant sein werden. Aktuell laufen nationale und internationale Kooperationen zur Anwendung des ncDHPR-Modells zur Klärung unterschiedlicher Krankheitsbilder.
(5. Juli 2021, Text: T. Mair, Foto: MUI/Institut für Pharmakologie, Grafik: eLife/Grabner, Dayal)
Publikationen:
Pore mutation N617D in the skeletal muscle DHPR blocks Ca2+ influx due to atypical high-affinity Ca2+ binding, DOI: 10.7554/eLife.63435
The mechanism underlying transient weakness in myotonia congenita, DOI: 10.7554/eLife.65691
Links:
https://www.i-med.ac.at/mypoint/news/713288.html
https://i-med.ac.at/mypoint/news/723869.html